The Division of Basic Research (DBR) explores fundamental scientific questions relevant to medicine and Anesthesiology.
Our research encompasses neurobiology, cell biology, and pharmacology with ongoing projects centered around the following areas: understanding mechanisms of hypoxic and immune-mediated cell death, investigating general anesthetic mechanisms, exploring ion channel function, identifying biomarkers of organ injury, uncovering novel intracellular signaling pathways, and studying the neurobiology of stress and addiction.
Our Team
The Division is led by Jose Moron-Concepcion, PhD, the Henry E. Mallinckrodt Professor of Anesthesiology, whose laboratory focuses on understanding the mechanisms underlying opioid addiction and the intersection with pain. Learn more about our team and labs below.
For general research inquiries, please contact the Research Administrative team.
PI Labs
The DBR is located within both the Neuroscience Building and the Clinical Sciences Research Building on Washington University School of Medicine’s campus. Principal investigators are members of the Division of Biological and Biomedical Sciences at Washington University and a number of graduate students have completed their thesis work in DBR labs.
Publications
Faculty in the Division of Basic Research continue to make meaningful contributions to research studies that span a range of topics.
Upcoming Research Events
Throughout the year, the Division of Basic Research hosts research seminars that offer opportunities for department members to become acquainted with ongoing research projects and engage in dialogue.
News
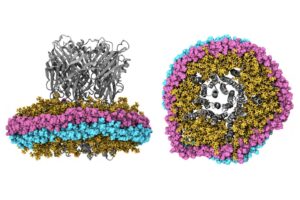
The Impact of Nanodiscs on Ion Channel Structure: Implications for Drug Design in Pain and Anesthesia
Moron-Concepcion named Mallinckrodt Professor of Anesthesiology

Dr. Vivian Gonzalez-Perez recruited as new tenure-track assistant professor
